Bartter, Gitelman & Liddle syndrome can be confusing for some students. In this post, these syndromes are simplified as much as possible to be fully understood.
Before starting reading the post, take some time to read this figure.
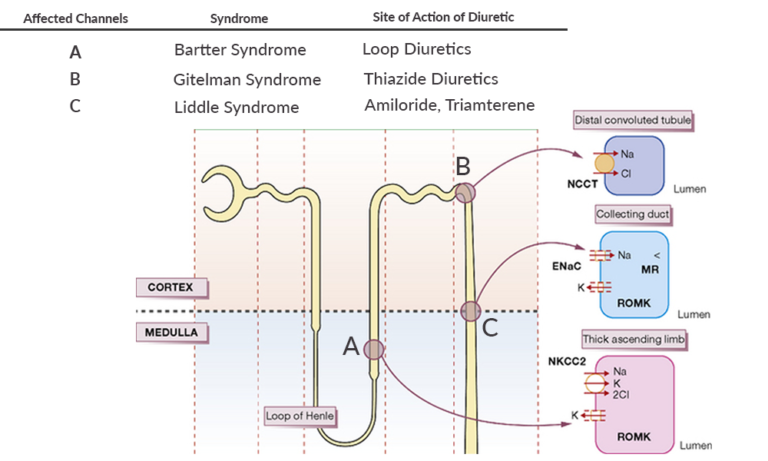
Bartter's syndrome
Bartter’s syndrome is an autosomal recessive disorder and is a major congenital cause of salt wasting.
Pathology
In this syndrome, the sodium-potassium 2 chloride (NKCC2) channel in the thick ascending loop of Henle is affected. There is a loss of function mutation in this channel, and so sodium and chloride reabsorption is defective leading to sodium & chloride with secondary water wasting. There is hypovolemia leading to activation of the renin-angiotensin-aldosterone system (RAAS). Increased sodium delivery from the loop of Henle to the collecting ducts results in increased sodium reabsorption in exchange for potassium and hydrogen ions, under the effect of aldosterone. This results in Hypokalemia and metabolic alkalosis.
Since the NKCC2 channel is also responsible for maintaining a gradient for Calcium reabsorption from the filtrate, therefore this reabsorption is also affected by this syndrome. Calcium is lost in the urine. This leads to hypocalcemia and hypercalciuria.
Clinical Features
Bartter syndrome presents in childhood with failure to thrive, polyuria, and polydipsia. Their blood pressure is normal.
Since the NKCC2 channel in the thick ascending loop of Henle is the site of action of loop diuretics, thus clinical and biochemical features in Bartter syndrome are similar to those in chronic treatment with Furosemide.
Treatment
Treatment is done with potassium replacement.
NSAIDs are used to counter prostaglandins present in this syndrome but they should be used after volume replacement.
ACE inhibitors can also be used in the treatment.
Gitelman's syndrome
Gitelman’s syndrome is also an autosomal recessive disorder.
Pathology
There is a loss-of-function mutation affecting the sodium chloride co-transporter (NCCT) in the early distal tubule. Just like Bartter syndrome, there is a loss of sodium and chloride along with water which results in secondary activation of the RAAS.
There is increased sodium delivery to collecting ducts, where under the effect of aldosterone, sodium is reabsorbed. Potassium and hydrogen ions are secreted and lost in the urine resulting in hypokalemia and metabolic alkalosis.
In this syndrome, there is also a defect in the function of the associated TRPM6 channel which is responsible for magnesium reabsorption. Therefore magnesium is also lost in the urine. So there results in hypokalemia, hypomagnesemia, and metabolic alkalosis.
Hypomagnesemia causes defective Parathyroid hormone secretion and results in hypocalcemia. Unlike in Bartter however, since the mechanism for hypocalcemia here is different so there is no hypercalciuria in Gitelman syndrome.
Clinical Features
The NCCT channel is also the site of action of thiazide diuretics. Therefore, the clinical and biochemical features of Gitelman syndrome are similar to chronic thiazide treatment.
Gitelman syndrome is milder in clinical features than Bartter syndrome. It presents in adolescence or adulthood with accidental findings of electrolyte abnormalities.
Treatment is with electrolytes replacement.
Liddle's syndrome
It is an autosomal dominant condition, affecting the Epithelial sodium channel (ENaC).
Pathology
There is a gain-of-function mutation of this channel, meaning that, unlike Bartter and Liddle syndrome, here the channel is hyperfunctioning. This hyperfunction leads to increased sodium reabsorption from the filtrate with secondary water absorption. This results in volume expansion and high blood pressure. Increased sodium reabsorption occurs at the expense of potassium and hydrogen ions loss, so there will be associated hypokalemia with metabolic alkalosis.
Treatment is with sodium restriction and potassium-sparing diuretics that block the ENaC channel, which is Amiloride or Triamterene.